Ist das menschliche Virom ein potenzieller Faktor der Infektionsanfälligkeit?
Worum geht es in diesem Forschungsprojekt?
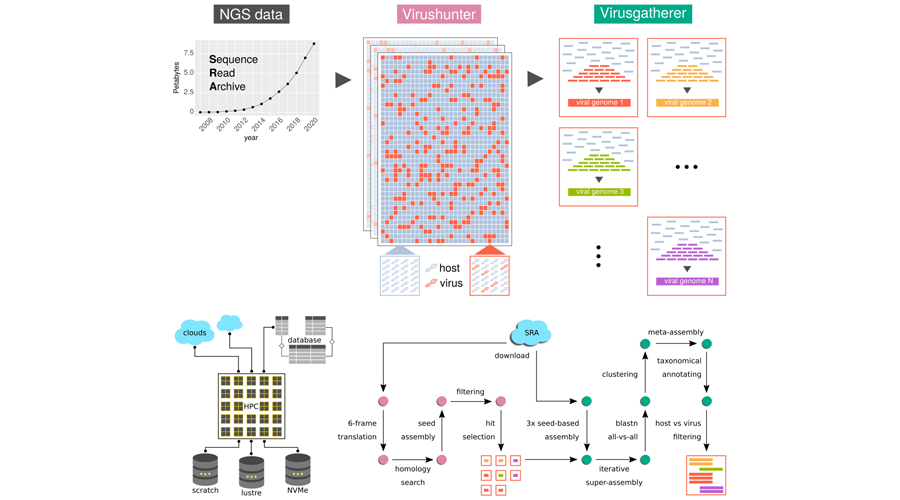
Bioinformatik-Workflow für die Entdeckung viraler Sequenzen in primären Sequenzierungsdaten unter Verwendung eines Hochleistungs-Computerclusters.
Worum geht es in diesem Forschungsprojekt?
Wir untersuchen die genetische Vielfalt der Virussphäre auf verschiedenen Ebenen, um die Vielfalt der Viren sowohl bei Eukaryonten als auch innerhalb des menschlichen Viroms besser zu verstehen. Mithilfe eines von uns entwickelten, auf Hochleistungscomputern basierenden Ansatzes zur Entdeckung von Viren suchen wir in Sequenzierungsdaten bekannte und unbekannte viraler Sequenzen, einschließlich hochgradig divergenter Viren ohne nahe Verwandte in Referenzdatenbanken. Wir wenden diesen Ansatz auf sehr große Mengen veröffentlichter Sequenzierungsdaten und auf unveröffentlichte Daten von Patientenkohorten an, um interindividuelle Unterschiede in der Zusammensetzung des Viroms zwischen kranken und gesunden Personen auszumachen.
Darüber hinaus versuchen wir, unseren Virusentdeckungsansatz auf Tierviren auszuweiten, um Primaten- und andere Wirbeltierarten zu identifizieren, die unbekannte Verwandte humanpathogener Viren beherbergen, was die Grundlage für die Entwicklung neuer Tierinfektionsmodelle bilden könnte.
Zudem sind wir am Projekt A1 beteiligt, in dem wir die genetischen Determinanten einer schweren Infektion mit dem humanen respiratorischen Synzytial-Virus (RSV) bei Säuglingen untersuchen.
Wie ist der Stand der Dinge?
Wir haben einen Hochleistungs-Computing-Workflow für die Entdeckung von viralen Sequenzen in unbearbeiteten Next Generation Sequencing (NGS)-Daten entwickelt. Diesen haben wir ursprünglich für die Suche nach neuartigen RNA-Viren in veröffentlichten NGS-Daten aus dem Sequence Read Archive (SRA)-Repository erarbeitet. Bislang konnten wir etwa 500.000 SRA-Datensätze screenen, die das gesamte Spektrum der verfügbaren eukaryotischen Transkriptome abdecken, dabei haben wir zahlreiche Sequenzen von bekannten und unbekannten RNA-Viren entdeckt.
Darüber hinaus haben wir etwa 76.000 menschliche SRA-Experimente mit verfügbaren Gewebe-/Organ-Annotationen analysiert, wodurch wir unter anderem zahlreiche bekannte und neuartige Anelloviren identifizieren konnten. Dabei war es uns möglich, häufig die Genome von Dutzenden von Viren in derselben Probe nachzuweisen. Das deutet darauf hin, dass innerhalb einer einzelnen Person virale Gemeinschaften existieren können. Diese Studien – und insbesondere die Analysen, die darauf abzielen, mögliche Zusammenhänge mit Gesundheit und Krankheit aufzudecken – sind noch nicht abgeschlossen.
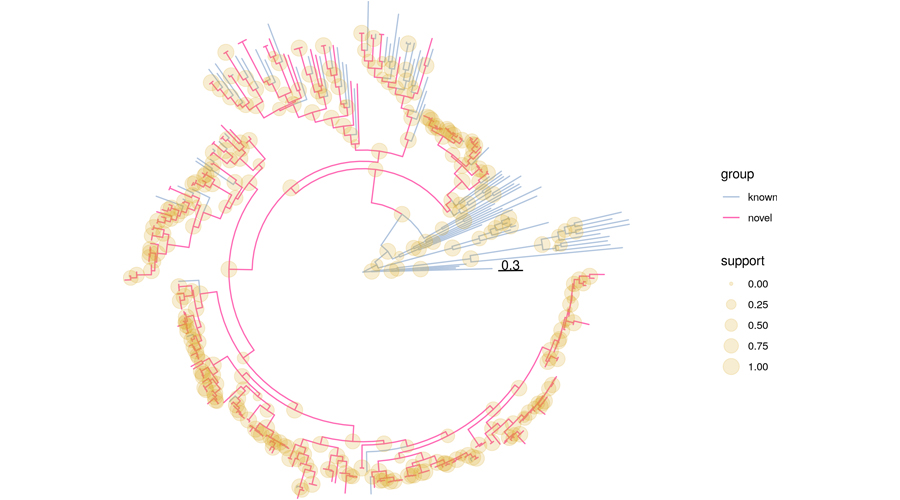
Phylogenetischer Baum der menschlichen und tierischen Anellovirus ORF1-Proteine.
Wie kommen wir da hin?
Wir werden unser Screening des SRA-Repository ausweiten, um viele weitere der Millionen von verfügbaren Humanexperimenten zu analysieren. Ergänzend dazu haben wir innerhalb von RESIST Kooperationen initiiert, um das Virom von Patientinnen und Patienten mit primären Immundefekten und von Frühgeborenen zu analysieren. Um die Empfindlichkeit unseres Ansatzes zur Entdeckung von Viren zu verbessern, haben wir mit der Entwicklung einer neuen Methode begonnen, die auf künstlichen neuronalen Netzen basiert. Wir beziehen Sequenzinformationen sowie Informationen über sekundäre und tertiäre Proteinstrukturen ein, die mit Methoden wie AlphaFold vorhergesagt werden. Wir erwarten, dadurch in der Lage zu sein, stark abweichende virale Sequenzen in NGS-Daten zu identifizieren, die in früheren ausschließlich auf Sequenzhomologie basierten Analysen unentdeckt blieben.
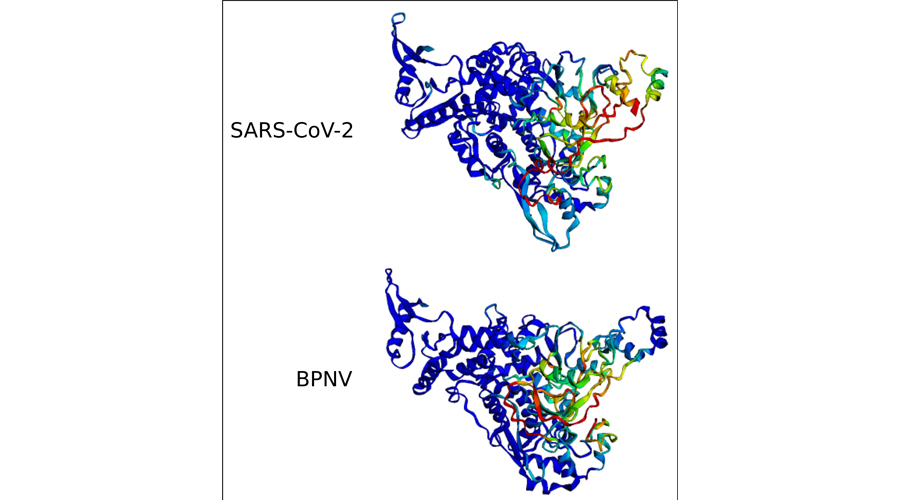
Proteinstruktur der viralen RNA-Polymerase von SARS-CoV-2 und des entfernt verwandten Ball-Python-Nidovirus.
