Is the human virome a potential factor of infection susceptibility?
What is this research project about?
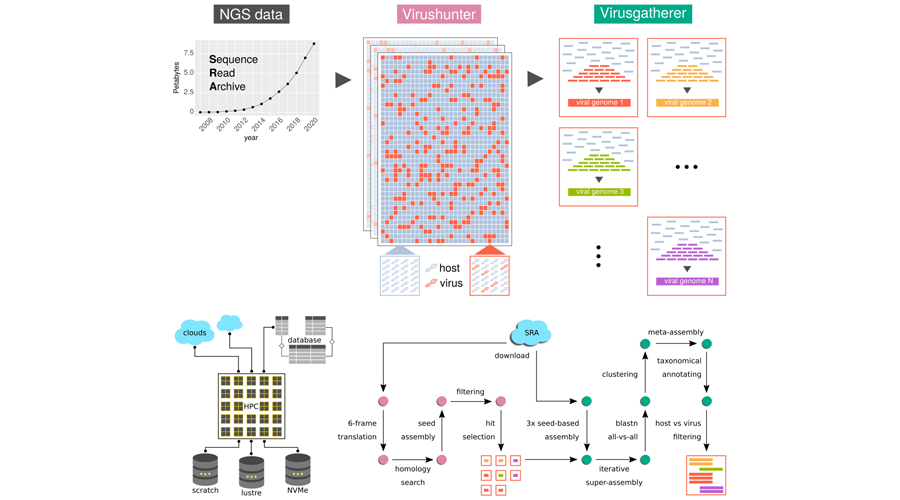
Bioinformatics workflow for discovering viral sequences in primary sequencing data using a high-performance computing cluster.
What is this research project about?
We study the genetic diversity of the virosphere at different scales because we are interested in better understanding the diversity of viruses both across eukaryotes and within the human virome. By utilizing a high-performance computing-based virus discovery approach developed by us, we screen sequencing data for the presence of known and unknown viral sequences, including highly divergent viruses with no close relatives in reference databases. We apply this approach to very large amounts of published sequencing data and to unpublished data from patient cohorts in order to explore inter-individual differences in virome composition between diseased and healthy persons.
Furthermore, we seek to extend our virus discovery approach to animal viruses, aiming to identify primate and other vertebrate species harboring unknown relatives of human pathogenic viruses, which may form the basis for establishing new animal infection models.
In addition to that, we are involved in project A1 where we study genetic determinants of severe infection with the human respiratory syncytial virus (RSV) in infants.
What’s the current status?
We have developed a high-performance computing workflow for the discovery of viral sequences in unprocessed next generation sequencing (NGS) data that we originally developed to search for novel RNA viruses in published NGS data from the Sequence Read Archive (SRA) repository. We have so far screened about 500,000 SRA datasets covering the full spectrum of available eukaryotic transcriptomes and discovered numerous sequences from known and unknown RNA viruses. In addition, we have analyzed about 76,000 human SRA experiments with available tissue/organ annotation, which led us to identify numerous known and novel anelloviruses, amongst others. Notably, we frequently detected the genomes of dozens of viruses in the same sample, suggesting that viral communities may exist within an individual person. These studies, and in particular analyses aiming to uncover possible association with health and disease, are ongoing.
What is this research project about?
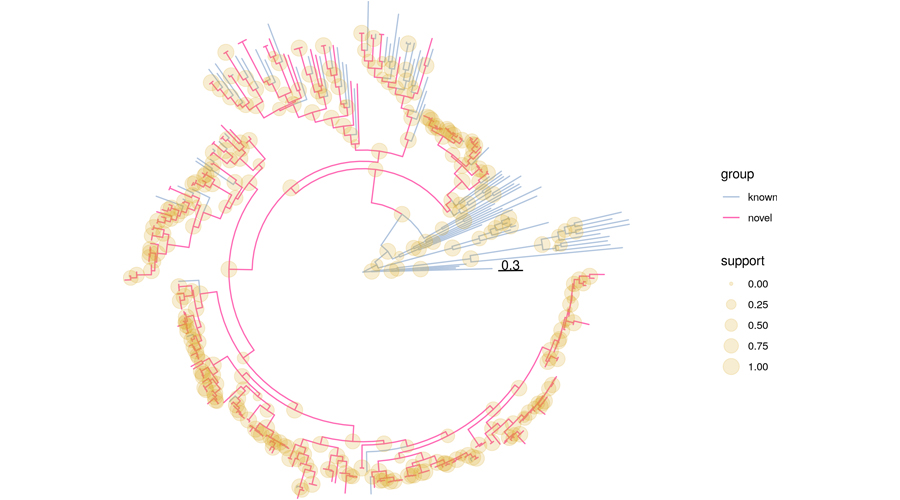
Phylogenetic tree of human and animal anellovirus ORF1 proteins.
How do we get there?
We will expand our screen of the SRA repository to analyze many more of the millions of available human experiments. Complementary to this, we have initiated collaborations within RESIST to analyze the virome of patients with primary immunodeficiencies and of premature infants. In order to improve the sensitivity of our virus discovery approach, we started with the development of a new method based on artificial neural networks. By incorporating both sequence information and secondary and tertiary protein structure information predicted by methods like AlphaFold, we expect to be able to identify highly divergent viral sequences in NGS data that remained undetected in previous analyses basing on sequence homology alone.
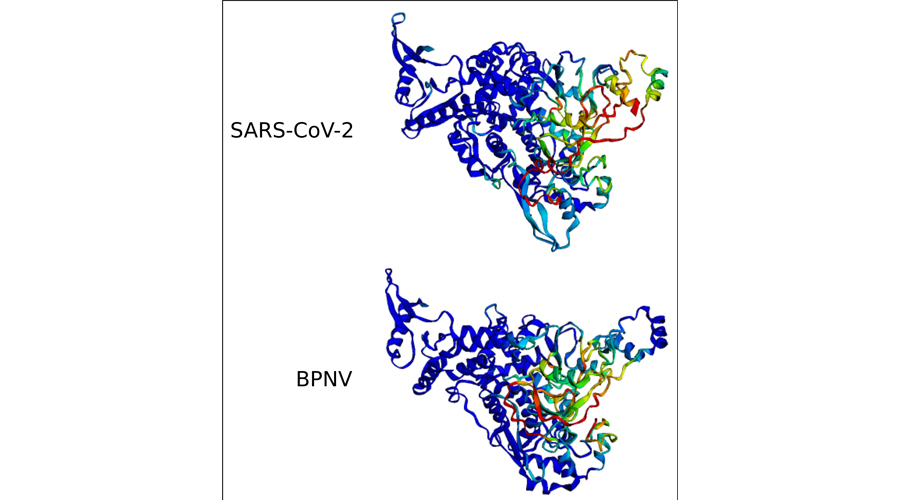
Comparison of protein structures of viral RNA polymerase of SARS-CoV-2 and remotely related Ball python nidovirus.
